医疗器械CE认证咨询
【服务简介】
CE是医疗器械产品出口欧洲的唯一路径,产品包装或产品本省加贴“CE”标志的前提是符合医疗器械法规REGULATION 2017/745(EU)的情况下,只有在符合法规要求的前提下加贴“CE”标志,产品才能合规上市。根据医疗器械法规REGULATION 2017/745(EU)(简称“MDR”)的要求,所有在欧盟境内上市销售的医疗器械产品必须符合MDR法规的要求并加贴CE标识,否则不得在欧盟境内上市销售。
欧盟MDR法规于2017年4月5日发布,由于受到新冠肺炎疫情的影响,原计划于2020年5月26日实施日期的MDR推迟至2021年5月26日。根据REGULATION(EU)2023/607的过渡期修订要求,在满足120.3c条款要求的情况下,在过渡期期间根据93/42/EEC或90/385/EEC颁发的证书,在未撤销的情况下,在证书到期后,最迟可以保持至2028年12月30日。
因此,在当前的法规要求下,对于未申请医疗器械CE证书的企业,只能根据器械类别所适合的符合性评估程序来申请符合MDR法规的要求。对于在过渡期期间根据93/42/EEC或90/385/EEC颁发CE证书的企业,在未撤销且120.3c的情况下,则可以选择继续保持原来的CE证书或选择根据MDR申请新的CE证书。
【服务内容】
1.MDR法规知识培训
2.医疗器械产品分类
3.辅导编写CE技术文件
4.辅导编写符合MEDDEV 2.7.1的临床评估报告
5.辅导符合MDR要求的质量管理体系
6.根据MDD指令要求编写技术文档
7.欧代和注册服务
8.Basic UDI-DI和UDI-DI的申请辅导
9.辅导企业进行审核整改
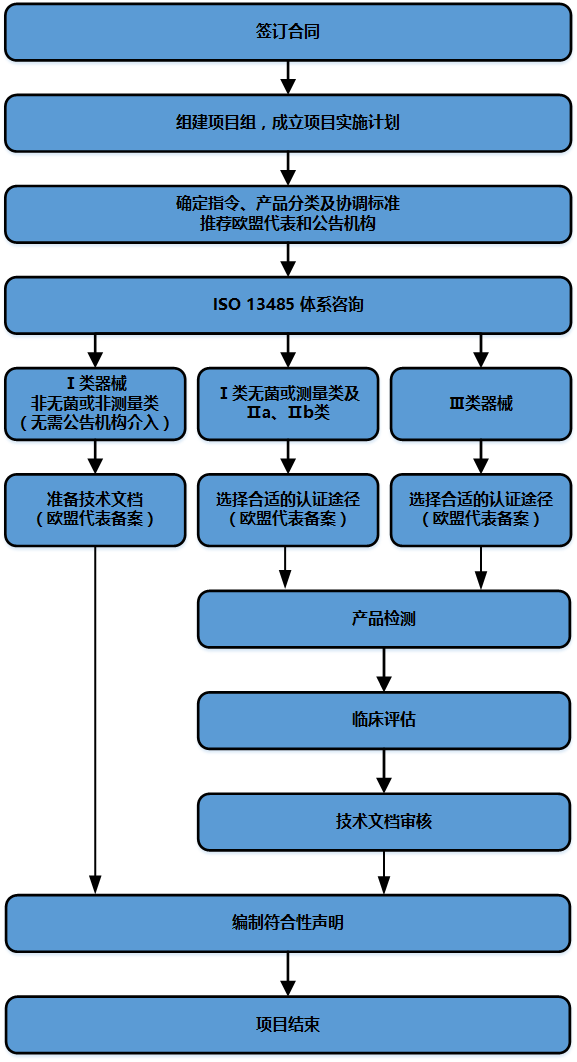
【服务流程】